
-
摘要:
缓蚀剂是用于防止金属材料腐蚀的化学物质,其有效性对于延长设备寿命、降低维护成本至关重要. 然而,传统的缓蚀剂分子筛选方法,如失重测量和电化学测试,通常需要大量实验和大量时间,成本高昂. 基于机器学习技术可以分析已知缓蚀剂分子数据,从而学习和预测新分子的缓蚀性能. 该方法可以提高筛选效率,揭示传统方法可能忽略的分子结构和性质,但其局限性也不容忽视. 首先,缓蚀剂分子筛选模型的化合物搜索空间有限. 其次,模型在实际应用中面临着与计算资源和时间成本相关的挑战. 在讨论了机器学习技术的应用和局限性之后,本文介绍了分子生成技术在发现新的高效缓蚀剂分子方面的应用以及挑战. 例如,生成模型需要大量高质量数据进行训练,生成的结果需要实验验证. 此外,生成模型在生成新分子时必须考虑分子稳定性、可合成性、环境影响等多种因素,使得模型的设计和优化更加复杂. 总体而言,机器学习技术在缓蚀剂分子研究中具有广阔的应用前景,但也面临着重大挑战. 通过不断优化机器学习算法并结合实验验证,有望在未来实现缓蚀剂分子的高效高精度发现,从而为材料科学和工业应用带来突破.
Abstract:In recent years, machine learning (ML) has demonstrated significant potential in corrosion inhibitor molecule research and has emerged as a powerful tool for scientists to explore new and efficient corrosion inhibitors. Corrosion inhibitors are chemical substances used to prevent the corrosion of metallic materials, and their effectiveness is crucial for extending equipment lifespan and reducing maintenance costs. However, traditional methods for screening corrosion inhibitor molecules, such as weight loss measurements and electrochemical testing, typically require extensive experiments and considerable time, making them costly. Consequently, the application of ML technology in this field has garnered widespread attention. This review provides an overview of the application of ML technology in screening corrosion inhibitor molecules. Artificial intelligence technologies, particularly deep learning and machine learning, can analyze vast amounts of data on known corrosion inhibitor molecules, to learn and predict the corrosion inhibition performance of new molecules. These technologies not only enhance screening efficiency but also uncover molecular structures and properties that traditional methods may overlook. Specifically, ML models can extract key information and construct predictive models through feature extraction and pattern recognition using existing data. These models can rapidly identify potential high-efficiency corrosion inhibitor molecules, thereby significantly accelerating research. However, despite the numerous advantages of ML technology in screening corrosion inhibitor molecules, its limitations cannot be ignored. First, the current compound search space for corrosion inhibitor molecule screening models remains limited. Second, these models face challenges related to computational resources and time costs in practical applications. After discussing the applications and limitations of ML technology, this study further explores the concept of molecular generation technology and its application in generating corrosion inhibitor molecules. Molecular generation technology employs deep learning techniques for automatically generating new molecular structures, often based on generative models such as generative adversarial networks (GANs) and variational autoencoders (VAEs). These technologies can learn the rules of molecular generation from existing corrosion inhibitor molecule data and generate new molecules with specific properties. Molecular generation technology can help researchers discover new and efficient corrosion-inhibitor molecules and accelerate the development of new materials. Finally, this paper highlights the challenges faced by generative machine learning models in the discovery of efficient corrosion inhibitor molecules. Although generative models have shown great potential for molecule generation and screening, their application in the discovery of corrosion inhibitors still faces many challenges. For example, generative models require large amounts of high-quality data for training, and the generated results require experimental validation. Moreover, when generating new molecules, generative models must consider various factors, such as molecular stability, synthesizability, and environmental impact, making the design and optimization of these models more complex. Overall, ML technology holds broad application prospects in the research on corrosion inhibitor molecules; however, it also faces significant challenges. Continuously optimizing ML algorithms and combining them with experimental validation should contribute to the efficient and high-precision discovery of corrosion inhibitor molecules in the future, leading to breakthroughs in materials science and industrial applications.
-
腐蚀是导致金属材料性能降低和结构破坏的主要原因之一,在世界范围内造成巨大的经济损失. 例如,李晓刚团队在2017年提到全球每年因腐蚀而损失的GDP高达2.5万亿美元,约占全球GDP的3.4%[1]. 减缓金属材料腐蚀的方法包括使用防护涂层、阳极保护、阴极保护、耐蚀合金设计、添加缓蚀剂等. 其中缓蚀剂是抑制金属腐蚀的一种有效方法,通过添加化学物质到腐蚀介质中,形成保护膜或改变腐蚀过程,从而减缓材料的腐蚀速率,具有成本低、操作简单、效率高等优点[2].
缓蚀剂的有效性通常通过缓蚀效率(Inhibition efficiency, IE)来评估,IE值越高表明缓蚀剂的腐蚀抑制效果越好,且与分子结构、浓度以及金属底物和腐蚀环境的变化密切相关[3]. 传统的实验评估IE的方法,如失重测量[4]、电化学测试[5],虽然广泛应用,但通常需要在多个实验条件下逐一测定特定浓度下缓蚀剂的IE. 这些方法不仅耗时且成本高昂,特别是在需要从广泛的化学空间中筛选出高效缓蚀剂及其合适浓度时,实验的高消耗和低效率成为了一大瓶颈. 除了实验方法,理论工具,如密度泛函理论(Density functional theory, DFT)计算和分子动力学(Molecular dynamics, MD)模拟,已广泛应用于缓蚀剂的研究[6−8]. DFT计算方法提供了缓蚀剂分子与金属表面之间电荷共享(Donor-acceptor)相互作用的重要信息[9],从而描述了缓蚀剂结构性质对腐蚀过程的影响[10−11]. 但是,DFT计算得到的量子化学参数众多,仅靠理论筛选缓蚀剂耗时且无法保证准确性. MD计算机模拟用于模拟缓蚀剂/表面系统,可视化吸附过程,并确定其相互作用的能量,以阐明介观水平(1到100纳米)、原子和分子水平上的缓蚀机制[12−16]. 但其计算成本高,且难以处理大规模的数据集,限制了其在广泛筛选中的应用.
随着人工智能技术的发展,机器学习(Machine learning, ML)尤其在缓蚀剂分子筛选、腐蚀机理研究[17−18]等领域显示出了巨大的潜力. ML算法通过分析大量已有数据,能够识别缓蚀剂分子特征与其缓蚀性能之间的复杂关系,从而挖掘出传统方法难以发现的规律. 与传统方法相比,ML不仅能够处理高维数据,还能综合分析多种因素(例如环境条件、缓蚀剂浓度、分子结构特征等)对缓蚀性能的影响. 因此,ML为缓蚀剂性能预测提供了一种高效、经济且创新的方法,推动了缓蚀剂研究和应用的发展. 例如多元线性回归分析[19−21]、神经网络[22−25]、支持向量机[26−27]、随机森林[28]、K-近邻算法[29]等机器学习技术[30−32]被应用于构建定量结构–活性/性能关系(Quantitative structure-activity/property relationships, QSAR/QSPR),从而建立缓蚀剂的IE与结构参数(电负性、极化率、范德华体积等)之间的相关性,以预测同系列分子的缓蚀性能. 然而,最近的文献表明,DFT衍生参数与IE之间的相关性具有误导性,或者对于大型缓蚀剂数据集来说,由于参数选择错误,或者计算方法选择错误,导致相关性太弱而无法定量分析[33−34]. 因此,已有作者借助消息传递神经网络(Message passing neural network, MPNN)等深度学习技术[35−36]建立分子结构与缓蚀剂分子的IE之间关系,可有效提高IE预测精度和模型泛化性能,从而快速筛选缓蚀剂.
但是上述方法均是从已有化学空间中筛选缓蚀剂,筛选效率过低,且无法生成新的缓蚀剂分子. 生成建模是机器学习的一个子领域,专注于开发能够生成新数据样本的算法,这些样本类似于给定训练数据集的数据分布,有期克服从超大型化学库中进行缓蚀剂筛选的局限性. 特别是条件式分子生成模型,可生成具有特定所需特性的分子,有助于生成特定腐蚀环境、特定缓蚀效率的缓蚀剂分子. 本文将首先对应用于缓蚀剂分子筛选的机器学习和深度学习技术进行全面分析,并讨论已有技术的局限性;然后简要介绍分子生成建模技术概念,给出其在高效缓蚀剂生成的可能应用以及挑战.
1. 缓蚀剂分子筛选方法
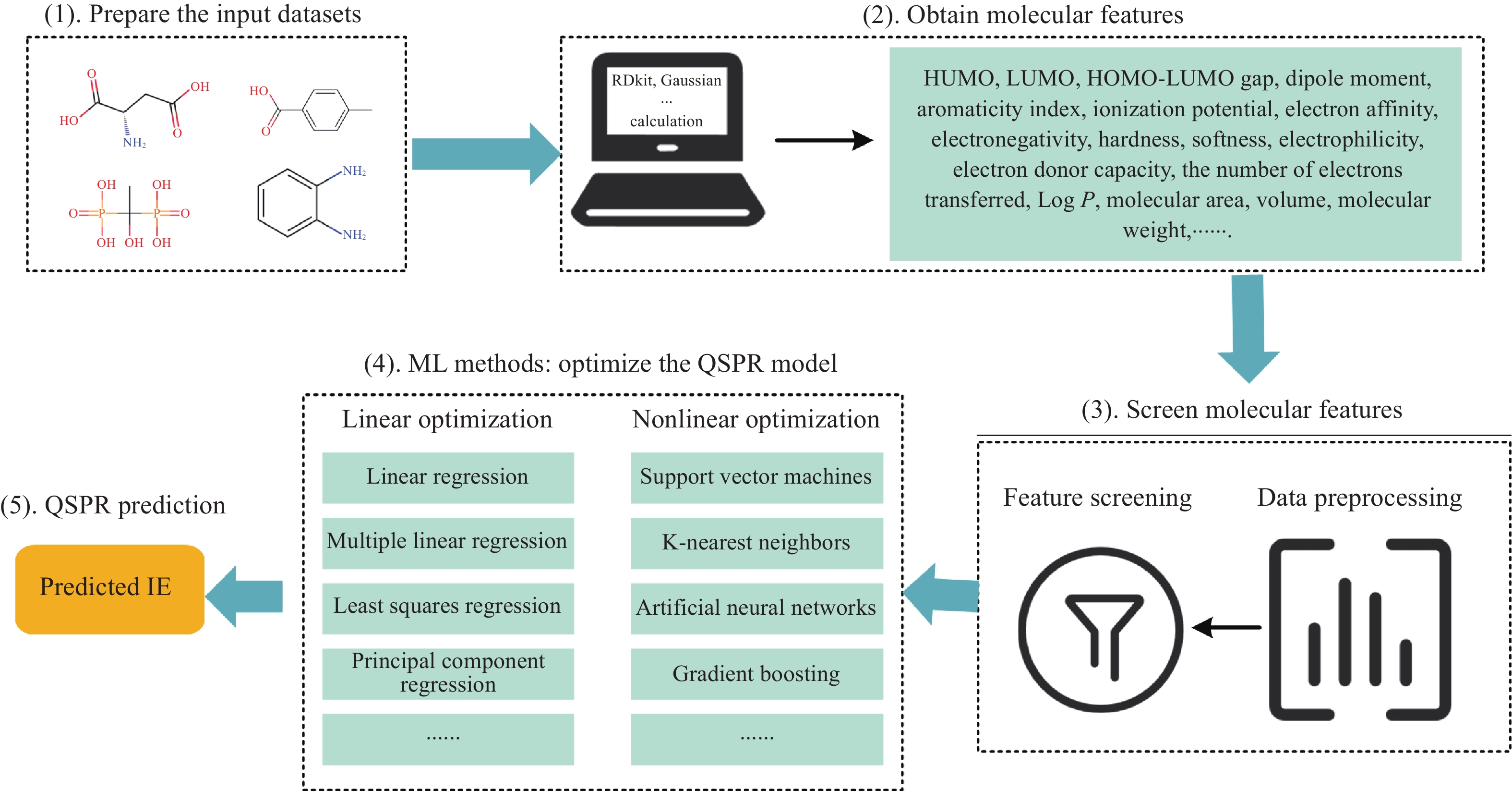
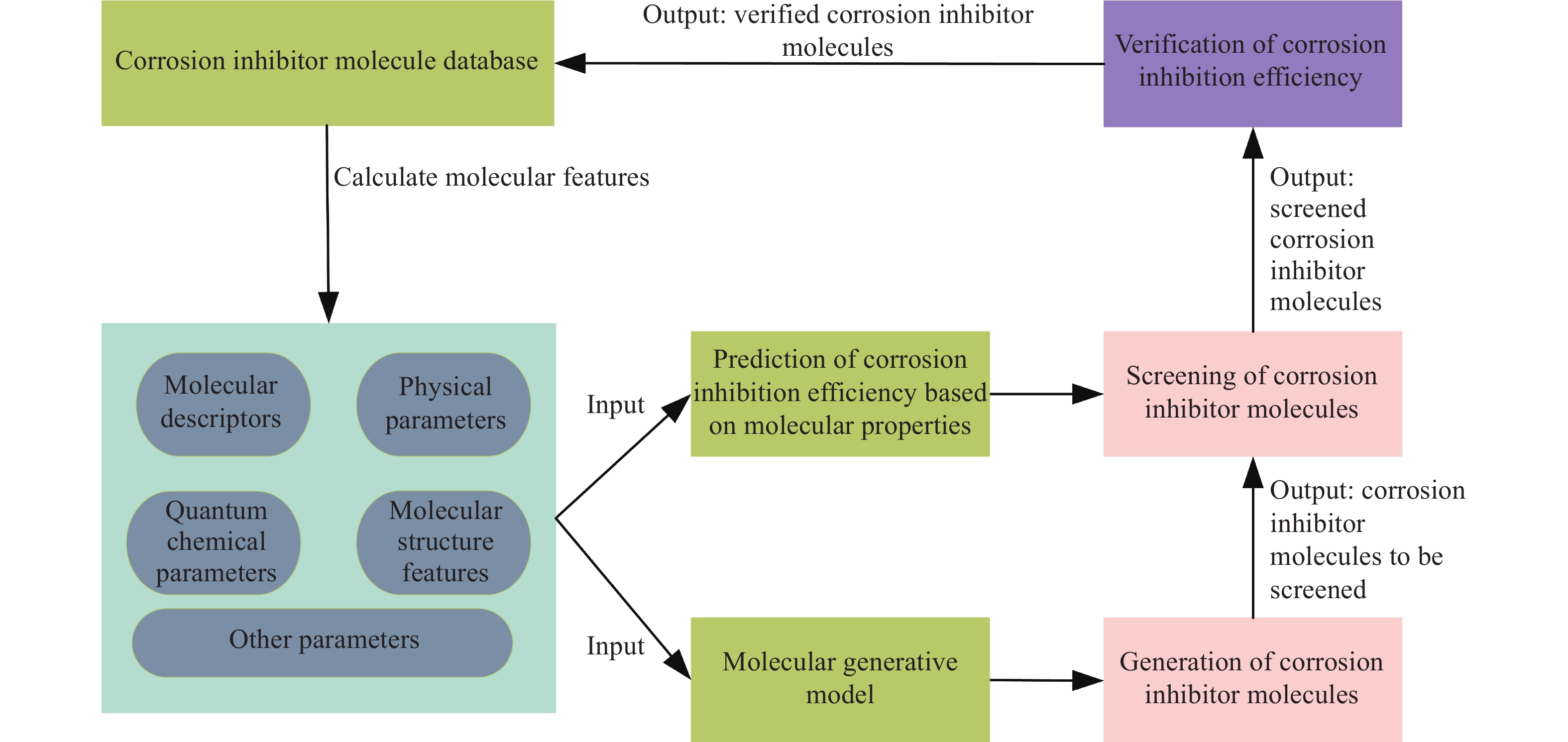
如图1所示,通过对已有缓蚀剂分子库进行分子特征计算,可分别作为IE预测模型和分子生成模型的输入,从而得到筛选后缓蚀剂分子,最后将经过实验验证的缓蚀剂分子放入缓蚀剂分子库中,此过程形成一个研究闭环. 其中,基于ML的缓蚀剂分子筛选主要以分子描述符、物理参数、量子化学参数或分子结构特征、以及环境因素(例如温度、环境pH值、缓蚀剂浓度等)作为预测模型的输入特征来预测缓蚀效率(IE). 常见的方法包括以分子描述符、物理参数、量子化学参数作为特征输入到ML模型进行IE预测的QSAR/QSPR分析,以及以分子结构特征作为输入进行机器学习或神经网络建模的IE预测方法.
![]() 图 1 ML助力缓蚀剂分子研究的思路Figure 1. Machine learning ideas for assisting research on corrosion inhibitor molecules
图 1 ML助力缓蚀剂分子研究的思路Figure 1. Machine learning ideas for assisting research on corrosion inhibitor molecules上述方法均通过建立分子特征(包括分子结构特征)与IE之间的关系,来实现缓蚀剂分子筛选,如图2所示,其建模步骤如下文列出.
![]() 图 2 面向IE预测的QSAR/QSPR建模流程Figure 2. Quantitative structure activity/property relationship (QSAR/QSPR) modeling process for inhibition efficiency (IE) prediction
图 2 面向IE预测的QSAR/QSPR建模流程Figure 2. Quantitative structure activity/property relationship (QSAR/QSPR) modeling process for inhibition efficiency (IE) prediction(1)准备输入数据集:收集并整理缓蚀剂分子特定环境(针对特定基体材料、腐蚀介质)下的IE数据、缓蚀剂浓度、介质浓度和分子特征数据.
(2)获取分子特征:IE预测输入的分子描述符主要包括利用RDkit软件包[37]计算得到的化学属性(例如分子量、辛醇/水分配系数(LogP)、拓扑分子极性表面积(TPSA)等)和使用化学信息学软件或工具(例如Dragon[38]、PaDEL-Descriptor[39]、CODESSA[40]、Gaussian、VASP、Materials Studio等)生成的计算参数,这些参数定量表示分子的结构特征、物理化学性质或基于分子结构得到的数据. 由于腐蚀抑制在很大程度上取决于分子在表面上的吸附能力[23],因此在缓蚀剂分子缓蚀效率预测算法的特征选择中,通常选择与控制吸附能力相关的参数,包括偶极矩、极化率、最高占位分子轨道(Highest occupied molecular orbital, HOMO)、最低未占分子轨道 (Least unoccupied molecular orbital, LUMO)、HOMO–LUMO间隙、电离势、电子亲和性、电负性、硬度、软度、范德华表面积、范德华体积等属性. 其中,分子的电子亲和性、电负性、硬度、软度等参数与分子的化学活性和反应性密切相关;HOMO–LUMO间隙反映了分子的稳定性和反应性,从而影响分子与金属表面的相互作用;偶极矩与分子的极性相关,影响其在金属表面的吸附能力[41].
(3)分子特征筛选:Rajan[42]在书籍《Informatics for Materials Science and Engineering: Data–Driven Discovery for Accelerated Experimentation and Application》中提到,分子特征的选择对于QSAR/QSPR建模至关重要,不恰当的分子特征的选择可能导致错误解释或者模型过拟合. 因此,分子特征的选择对于QSAR/QSPR建模精度起到至关重要的作用,可以通过专家经验、统计学方法或机器学习算法,筛选出与目标性质相关性较高的分子特征,去除冗余和无关的特征,提高模型的准确性和效率.
(4)QSPR模型优化:选择适当的线性优化(如多元线性回归)或非线性优化的机器学习算法(如人工神经网络),并通过交叉验证和参数调优等方法优化模型性能,确保模型的泛化能力和预测精度. 一般常利用均方误差(Mean squared error, MSE),均方根误差(Root mean square error, RMSE)和拟合优度指标$ R^{2} $几个指标评估QSPR模型性能并进行验证.
(5)QSPR模型预测:使用优化后的模型对新分子进行性质预测.
由于特征工程和模型优化两个部分对于IE预测精度影响较大,因此,第2节和第3节将针对两部分进行综述.
2. 特征工程−分子描述符筛选
在进行IE预测模型构建过程中,特征工程严重影响了模型的性能. 特征工程主要包括数据预处理、特征选择、特征降维和特征构建几个方面.
2.1 数据预处理
数据预处理是确保机器学习模型能够有效学习和预测IE的重要步骤. 数据预处理的主要步骤和方法包括:
(1)数据清理. 主要包括缺失值处理、异常值处理. 其中缺失值处理可通过删除法、插值法(例如线性插值)、填充法(例如均值、中位数、众数进行填充)、模型预测法预测缺失值. 异常值处理可通过使用箱线图、z-score等统计方法识别和处理异常值,或使用孤立森林和局部离群因子(Local outlier factor, LOF)[43]等算法检测异常值,或结合领域知识手动检查和处理异常值.
(2)数据转换. 在进行模型训练和测试前,输入变量之间可能存在量纲上的差别,为了尽可能减少量纲差别对各个输入特征的影响,可采用z-score标准化、min–max归一化、标签编码、独热编码(例如不同缓蚀剂分类、不同腐蚀介质分类)等方式对数据进行转换,以保证模型能正确理解这些特征.
(3)数据增强. 由于ML建模需要较大的数据量,而缓蚀剂数据较少,因此可通过数据增强的方法对数据集进行扩充. 例如Sutojo等[29]为了克服缓蚀剂化合物数据集的小样本问题,使用虚拟样本生成(Virtual sample generation, VSG)方法生成虚拟样本并将其添加到训练集. 利用六个小数据集验证了在训练数据中加入虚拟样本,有助于KNN算法识别特征–目标关系模式,从而增加与缓蚀效率相关的量子化学描述符的数量.
(4)数据拆分. 在ML模型建立中,需要按比例随机划分训练集和测试集数据集,如80%用于训练,20%用于测试. 也可采用如k折交叉验证,通过多次训练和测试确保IE预测模型的稳定性和泛化能力.
2.2 分子特征筛选
在IE预测建模过程中,常见的用于对分子描述符进行特征选择、特征构建、特征降维或者回归建模的统计分析和ML方法主要包括主成分分析法(Principle component analysis, PCA)[41]、遗传算法(Genetic algorithm, GA)[21,23,44]等方法.
(1)主成分分析(PCA)是一种常用的无监督降维技术,旨在通过线性变换将高维数据投影到低维空间,同时保留尽可能多的数据方差. PCA通过找到数据中方差最大的方向作为主成分来实现降维. 将高维的分子特征数据降维到较低维度,可更好地进行可视化、建模或分析. 通过选择最重要的主成分来构建新的特征,用于后续的回归任务. 例如Hadisaputra等[41]对HOMO、LUMO、HOMO–LUMO间隙、偶极矩、电离电位、电子亲和力、硬度、软度、电负性、电子转移分数、亲电性指数、LogP、临界体积和相对分子质量几个分子特征进行PCA分析,研究呋喃衍生物缓蚀剂对低碳钢的腐蚀抑制性能,得出如下结论:LUMO、HOMO–LUMO间隙、偶极矩、软度、电子转移分数、亲电性指数和反馈能对主成分1有显著贡献;相对分子质量和临界体积对主成分2有显著贡献;HOMO、电离电位和 LogP 对主成分3有显著贡献.
(2)遗传算法(GA)是一种模拟生物进化过程的优化算法,通过模拟自然选择、交叉和变异等过程来搜索问题的解空间. 在特征选择或特征优化中,遗传算法可以用来探索最优特征子集的组合. 例如Ser等[23]利用GA研究了酸性介质中,41种吡啶和喹啉N-杂环化合物对铁合金的缓蚀性能影响因素. 通过结合人工神经网络和GA建模得出最优九个变量(HOMO、LUMO、HOMO–LUMO 间隙、电负性、柔软度、亲电性、电子供体容量、N原子电荷和吸附能指数),揭示了吸附能、物理尺寸等参数对金属腐蚀抑制的重要性,提出了关键的腐蚀抑制设计原则.
3. 面向IE预测的模型优化与预测
根据模型输入是分子特征还是分子结构进行分类,可将建模划分为QSAR/QSPR模型构建和基于分子结构的IE预测模型构建.
3.1 QSAR/QSPR模型构建
基于分子特征进行QSAR/QSPR模型构建方法可分为三大类,即线性优化方法(多元线性回归和偏最小二乘回归)、非线性优化方法(人工神经网络、支持向量机、K最近邻算法、决策树、随机森林等)以及混合优化方法. 通过调研已有IE预测的QSAR/QSPR模型,发现主要采用多元线性回归[20−21,44−47]、人工神经网络[24,46−48]、梯度增强[27,31,41,49−50]几种方法,输入的分子特征主要为物理参数和量子化学参数.
支持向量机(Support vector machine,SVM)是一种基于结构风险最小化原理的统计学习方法. 近年来,它已成功应用于解决众多领域的分类和回归问题,并在实际应用中提供最先进的性能. 支持向量机的预测结果具有快速学习、全局优化和出色的泛化能力等诸多优点. 另一方面,SVM在小样本数据集上表现出色. Zhao等[27]针对在 1 mol·L−1 盐酸中,浓度为 0.01 mol·L−1的氨基酸对铁腐蚀的动电位极化曲线获得的IE数据,基于SVM模型,以HOMO、LUMO、偶极矩(μ)、电离势(I)、电子亲和力(A)、电负性(χ)、硬度(η)、软度(σ)、分子体积(V)、转移电子分数(ΔN)、自然电荷(Qtotal)、Mulliken电荷(Ztotal)等物理化学参数作为模型输入,构建了非线性QSAR模型,模型预测和实验IE之间的差异(RMSE)为1.48. 为了对比不同阶段的相关程度,给出在气相和水相条件下的相关系数绝对值(|R|),发现|R|均小于0.3,表明氨基酸的缓蚀性能与气相和水相中单个量子化学参数的相关性较低.
多元线性回归(Multiple linear regression, MLR)是一种用于建立因变量与多自变量之间线性关系的模型,一般采用最小二乘法,即最小化残差平方和来估计模型参数. 由于MLR模型相对简单,回归系数直接反映了每个自变量对因变量的影响,易于理解,计算复杂度较低,适合处理大规模数据集,且可以通过加入交互项和非线性项扩展到更复杂的模型. 因此,MLR广泛被应用于QSAR/QSPR建模,用于预测和解释多个分子描述符因素对IE变量的共同影响. 例如,Quadri 等[24]用20 种吡哒嗪衍生物在酸溶液中对低碳钢的缓蚀效果进行了评价. 根据化合物的化学结构和电子结构的不同,IE从65%到97%不等. 包括HOMO–LUMO能隙、HOMO、LUMO、μ、I、A、χ、η、σ、ΔN、总能量(TE)在内的分子的结构性质,由GaussView 5.0 查看结构后使用 Gaussian进行几何优化进行确定. 利用缓蚀剂浓度(Conc)、温度(Temp)、实验参数和理论参数建立了一个MLR模型来预测观察到的IE,MSE、RMSE和平均绝对百分比误差的预测结果分别为
111.5910 、10.5637 和10.2362. IE与常规键级之和(Sum of conventional bond orders, SCBD)、软度(σ)、LUMO、电离势(Mi)和分子量(MW)密切相关. 但MLR也存在一定局限性,例如(1)MLR假设因变量与自变量之间的关系是线性的,因此无法捕捉非线性关系;(2)虽然模型简单,但在高维空间中解释性可能下降,尤其当自变量数量较多时,模型容易过拟合. 因此,需要通过适当的数据预处理和模型改进,克服一些局限性,提高模型的适用性和预测能力.人工神经网络(Artificial neural network, ANN)是一种模拟生物神经网络行为的计算模型,由大量的人工神经元(节点)组成,这些节点通过加权连接相互作用. ANN通过学习和调整权重来从输入数据中提取复杂的模式和非线性关系. 在建立分子描述符与IE关系的QSAR/QSPR模型中,ANN能够处理高度非线性和复杂的数据关系,理论上可以逼近任意复杂的函数. 具体过程包括:将分子描述符作为输入层节点,通过多个隐藏层进行非线性变换,最后在输出层得到预测的IE值. 通过反向传播算法(Backpropagation),调整网络权重以最小化预测误差,从而实现精确的QSAR/QSPR模型. 例如Ser等[23]以9个输入分子描述符(计算吸附能指数、HOMO、LUMO、HOMO–LUMO 能隙、电负性、柔软度、亲电性、电子供体容量和N原子电荷)作为输入,建立基于遗传算法–人工神经网络(GA–ANN)方法的QSPR模型. 通过与基于遗传算法–偏最小二乘(GA–PLS)的QSPR模型进行性能对比,结果显示GA–ANN方法在训练集、测试集和验证集上的平均RMSE优于线性的GA–ANN方法,得到的平均RMSE(训练/测试/验证)为8.8%. 但ANN也存在一定局限性,例如:(1)数据量不足时容易导致模型过拟合或欠拟合;(2)可能会遇到梯度消失或梯度爆炸问题,尤其在深层网络中,这会导致训练不稳定或难以收敛;(3)神经网络的“黑箱”性质使得很难解释模型的内部机制和预测结果.
梯度增强(Gradient boosting, GB)是一种迭代的集成学习方法,通过逐步构建多个弱学习器(通常是决策树)来优化模型性能. GB通过决策树的分裂过程,可自动选择重要特征,减少手动特征选择的工作量,且相比ANN更易于解释. 另一方面,由于逐步减少残差的过程,梯度增强方法对噪声数据具有一定的鲁棒性. 例如,Akrom等[31]设计了包含50种天然有机化合物的数据集,以11种量子化学性质(HOMO、LUMO、HOMO–LUMO能隙、电离势、电子亲和力、全局硬度、全局柔软度、电负性、偶极矩、亲电性、转移电子分数)和化合物浓度作为输入特征,以IE值作为目标变量. 为了提高机器学习模型的预测精度,在训练过程中使用核密度估计函数生成虚拟样本,并测试了k最近邻(K-Nearest neighbors, KNN)、随机森林(Random forest, RF)和GB三种不同的模型的IE预测性能. 结果表明,由于引入了虚拟样本,有效地提高了输入特征与目标值之间的相关性,模型的预测性能得到了显著提高. GB、RF和KNN模型的R2值分别从0.557增加到0.996、0.522增加到0.999、0.415增加到0.994. 此外,每个模型都显示RMSE值的显著降低,分别从1.41过渡到0.19、1.27过渡到0.10和1.22过渡到0.16. 可见,基于GB方法构建的模型更有利于该数据集进行IE准确预测. 但GB方法也同样有一定局限性,例如:(1)GB方法涉及多个超参数(如学习率、弱学习器数量、树的深度等),需要仔细调优才能达到最佳性能;(2)在弱学习器数量过多或模型过复杂时,GB方法容易发生过拟合,需要使用正则化方法(如缩减树的深度、增加学习率衰减等)来防止过拟合.
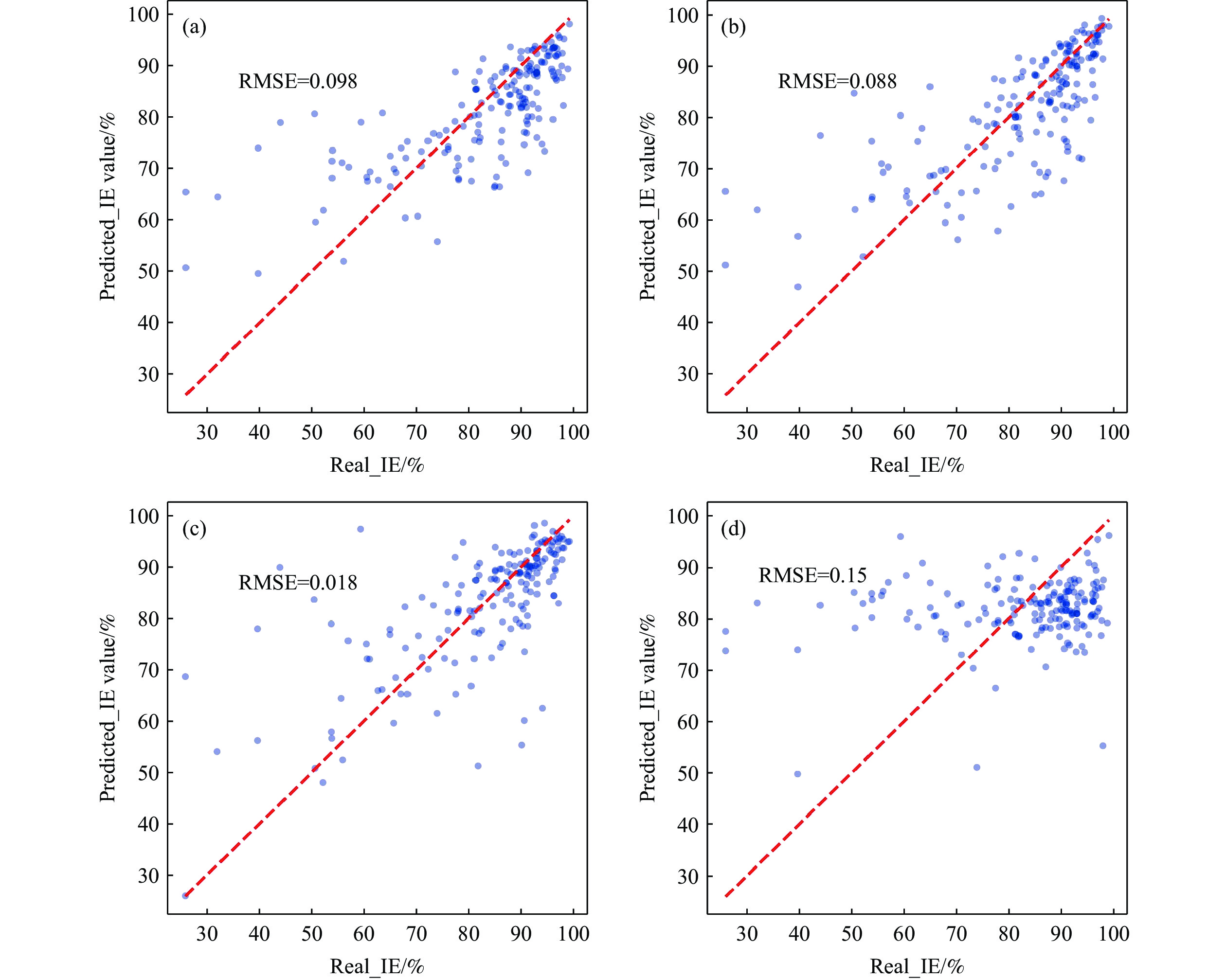
上述方法各有特点,适用于不同的数据集和预测需求,但都展示了在缓蚀剂分子筛选中的潜力和应用前景. 通过结合适当的数据预处理和正则化方法,选择合适的模型和分子描述符,可以有效地建立分子描述符和缓蚀性能之间的关系,为实验和理论研究提供了重要的支持和指导. 为了对比上述四个模型对于缓蚀剂分子的缓蚀效率(IE)预测准确度,本文采用随机森林(RF)、梯度增强(GB)、人工神经网络(NN)和多元线性回归(MLR)四个算法对Gong等[51]提供的特定环境条件下(环境温度:25 ℃~ 30 ℃,HCl浓度:1 mol·L−1)的缓蚀剂分子数据集(包括缓蚀剂浓度、HOMO、LUMO、偶极矩、电离电位、电子亲和力、硬度、软度、电负性、范德华体积、范德华表面积等特征参数)进行IE预测模型构建. 为确保实验的公平性,四个模型均采用8∶2的比例将数据集划分为训练集和验证集,并通过十折交叉验证进行模型训练. 如图3所示,NN模型在验证集上的RMSE为0.018,表现出最高的准确度;RF和GB模型的表现相近,而MLR模型的准确度最低,RMSE为0.15. 因此,针对缓蚀效率的预测,NN、GB和RF模型展现出更优的预测准确性.
![]() 图 3 不同模型预测缓蚀剂在1 mol·L−1盐酸溶液中的缓蚀效率准确度对比图. (a) 随机森林模型; (b) 梯度增强模型; (c) 人工神经网络模型; (d) 多元线性回归模型Figure 3. Prediction accuracies compared for corrosion inhibition efficiency of inhibitors in 1 mol·L−1 hydrochloric acid solution, using different models: (a) random forest; (b) gradient boosting; (c) artificial neural network; (d) multiple linear regression
图 3 不同模型预测缓蚀剂在1 mol·L−1盐酸溶液中的缓蚀效率准确度对比图. (a) 随机森林模型; (b) 梯度增强模型; (c) 人工神经网络模型; (d) 多元线性回归模型Figure 3. Prediction accuracies compared for corrosion inhibition efficiency of inhibitors in 1 mol·L−1 hydrochloric acid solution, using different models: (a) random forest; (b) gradient boosting; (c) artificial neural network; (d) multiple linear regression3.2 基于分子结构的IE预测模型构建
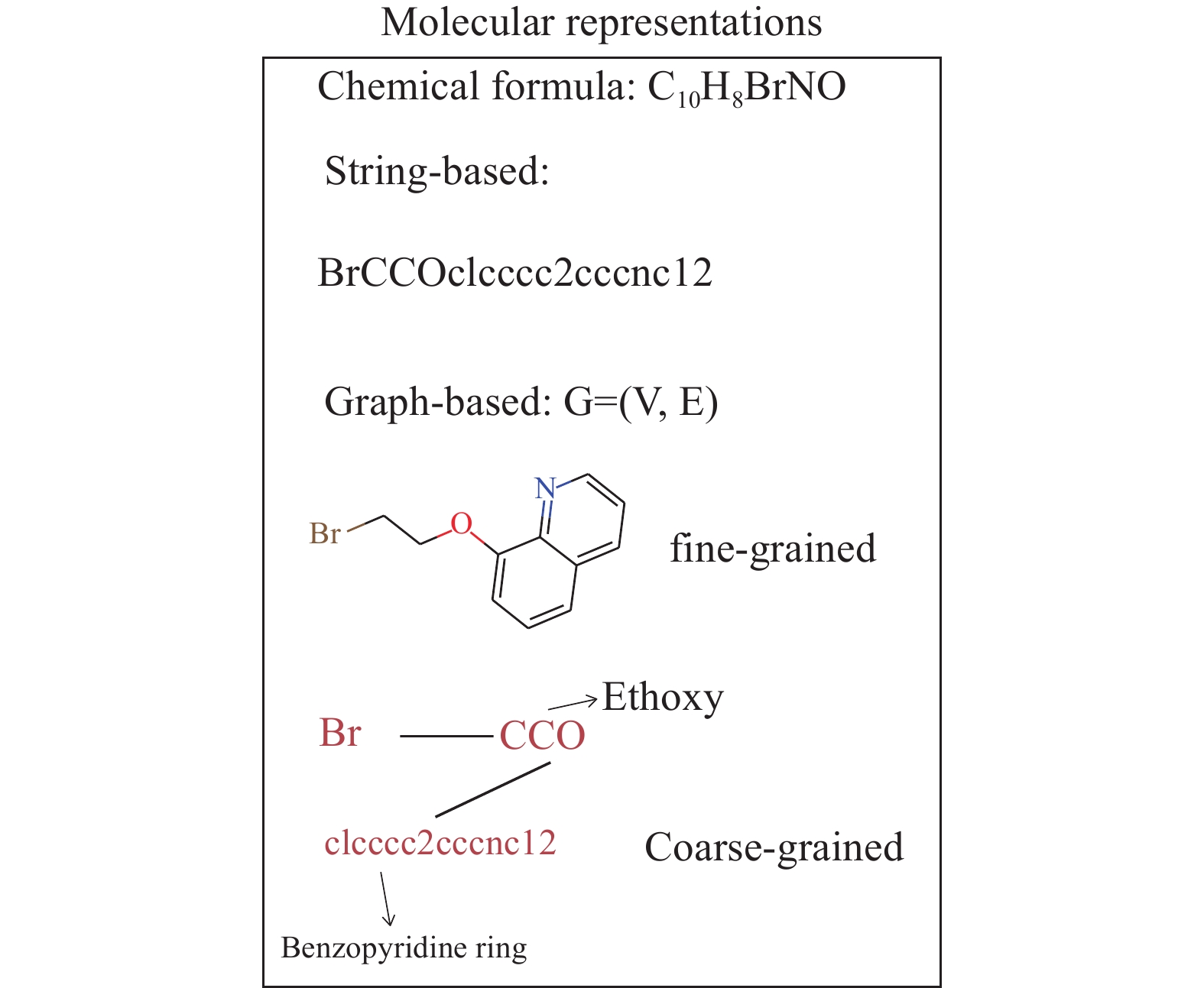
在基于分子结构的IE预测建模中,需要对缓蚀剂分子进行分子表示后进行建模. 分子表示方法包括以字符串进行编码的SMILES形式、分子指纹、图的形式. SMILES格式[52]基于一定规则的语法词典进行编码,适用于循环神经网络、transformer等自然语言模型. 分子指纹一般采用摩根编码为
1024 或2048位的0、1向量. 令分子图表示为G=(V, E),则G的表示按照分子粒度区分,细粒度表示可以以原子为节点(V),化学键为边(E),可精确描述每个原子和键的变化;粗粒度表示将分子分割为多个片段或官能团,以片段或官能团为节点,连接关系为边来构造图数据,可以简化输入数据,提供ML模型训练速度和效果. 例如图4中SMILES为BrCCOc1cccc2cccnc12的缓蚀剂8-(2-bromoethoxy)quinoline(QN-C2Br),按照Br, Ethoxy和Benzopyridine 环三个片段作为节点可得到粗粒度的图结构.传统的深度卷积神经网络(Convolutional neural network, CNN)和递归神经网络(Recurrent neural networks, RNN)只能处理文本、音频、图像、视频等欧几里得数据. 与图像和文本不同,图数据包含基本的结构信息. 图神经网络(Graph neural network, GNN)是直接从由节点和边组成的图数据中学习的框架,其中节点和边可以很好地用于表示分子结构中的原子和键[53]. 因此,GNN可用于处理非欧几里得数据,如化学分子结构和蛋白质,并从其结构预测分子的性质[54],包括量子力学特征,如能量、电子和热力学性质[55−57];理化性质,如疏水性、水中的水合自由能、辛醇/水分配系数(LogP)[58]和毒性[59].
缓蚀剂的缓蚀效率不仅与其内部结构参数(如杂化程度、每个原子的键数、每个原子的价电子数和键类型)密切相关,而且与整体分子特征(如分子量、芳环数、受体数和给体数)密切相关. 消息传递神经网络(Message passing neural network, MPNN)是监督图学习的通用框架,它简单地抽象了几种最有前途的GNN模型之间的共性,能够直接从分子图中学习原子级和化学键级特征,并预测分子性质. 因此,Dai等[35]通过检索116篇以“缓蚀剂”为关键词研究盐酸溶液中不同缓蚀剂分子对碳钢影响的论文,得到特定环境条件下(环境温度:25 ℃~ 30 ℃,缓蚀剂浓度:1 mmol·L−1,HCl浓度:1 mmol·L−1)的缓蚀剂名称、类别、分子结构和实验IE值,共270条数据. 基于该数据,Dai等[35]提出了一个基于DMPNN框架的三级直接消息传递神经网络(3L-DMPNN)模型,通过结合原子水平特征、化学键水平特征和分子水平特征来筛选缓蚀剂. 具体而言,使用简化分子输入行输入系统(Simplified molecular-input line-entry system, SMILES)[52]作为唯一输入,将分子结构视为图,并使用开源的RDKit包从SMILES中提取原子和化学键特征[39]. 随后,将消息传递模块后的新分子图向量与全局分子特征相结合,通过前馈神经网络预测分子的IE. 而Ma等[36]在3L-DMPNN模型的基础上提出2D3DMol-CIC模型,对数据集进行扩充,得到包含缓蚀剂名称、分子的SMILES、分子中的原子坐标、缓蚀剂浓度和IE值的数据
1241 条,然后结合2D–3D分子图和缓蚀剂浓度进行建模,验证了三维特征对IE预测的影响以及模型的泛化能力. 与已有的预测模型相比,所构建的2D3DMol-CIC模型不仅可以更准确地预测特定浓度下跨类缓蚀剂的IE,而且可以预测不同浓度下缓蚀剂的IE,从而确定产生高IE(>90%)的最小浓度.4. 局限性分析与展望
虽然通过提取定量分子描述符等结构化数据,可以建立IE预测模型,在几分钟甚至更短的时间内预测IE,从而基于IE进行缓蚀剂分子筛选. 然而仍存在以下问题:(1)基于ML建立的QSPR模型在很大程度上依赖于分子特征的选择,仅限于预测特定浓度下某类缓蚀剂的IE;(2)目前已有文献[60]提到电离势、HOMO或LUMO能量或任何其他量子化学衍生的描述符与腐蚀效率之间基本上没有相关性,因此,利用量子化学参数作为IE预测模型输入参数是否准确有效,仍有待研究;(3)利用深度学习技术,建立基于分子结构的IE预测模型可一定程度提高IE预测精度和模型泛化性能,能较准确地预测缓蚀剂及其浓度,为缓蚀剂及其浓度的筛选提供了一种低成本、快速的方法,但是仍然只能在有限的化合物空间进行搜索.
在未来研究中,一方面,可尝试利用高通量计算技术,生成大批量缓蚀剂分子,并利用神经网络模型建立基于分子特征的缓蚀效率预测模型;另一方面,基于分子生成模型生成新的缓蚀剂分子有可能克服从大型化合物空间进行缓蚀剂分子搜索的局限性. 例如利用条件生成模型可按照特定属性进行分子设计,从而缩小化合物搜索空间. 目前,在药物发现领域,已有研究利用生成式建模得到现有化学库以外的、经过实验验证的化合物[61−62]. 针对SMILES、graph两种常用分子表示方法的分子输入数据,分子生成模型可主要分为自回归模型(Autoregressive models)、生成对抗网络(Generative adversarial networks, GANs)、变分自编码器(Variational autoencoders, VAEs)和规范流模型(Normalizing flows, NFs)四类. VAEs包括一个经过训练的编码器,用于参数化潜在变量z的分布,以及一个经过训练的解码器,用于使用编码器定义的分布中的样本重建输入,因此适用于缓蚀剂分子生成建模. 在缓蚀剂分子生成方面,Gong等[51] 利用Ma等[36]文献提供的基于碳钢材料的
1368 条缓蚀剂数据,基于RDKit软件计算了包括药物相似性定量估计(Quantitative estimate of drug-likeness, QED), 分配系数的对数(ALOGP, LogP), 分子量(Molecular weight, MolWt), 氢键受体(Hydrogen bond acceptors, HBA), 氢键供体(Hydrogen bond donors, HBD), 拓扑极性表面积(Topological polar surface area, TPSA), 可旋转键数量(Number of rotatable bonds, NumRotBonds)和N、O、S、P原子数量11个分子特征,以这11个分子特征和缓蚀剂浓度作为缓蚀剂分子生成模型的输入条件,建立条件式VAEs模型. 如图1所示,基于生成模型生成的新分子可作为缓蚀剂分子筛选的候选集,然后利用IE预测模型得到筛选后的缓蚀剂分子,从而减少缓蚀剂分子筛选的化合物空间.尽管已有众多可供选择的分子生成方法来针对缓蚀剂分子进行分子生成模型训练,但在缓蚀剂分子生成领域仍存在较多的问题亟待解决.
(1)不同缓蚀剂针对不同基体材料、不同腐蚀环境所体现的缓蚀效率不同,因此,在进行缓蚀剂分子生成建模时,需要针对特定腐蚀环境、特定基体材料,收集相关缓蚀剂分子数据,但是目前已有数据集稀少,可能导致生成有效分子的比例过低. 因此,需要利用文献挖掘、高通量实验等方式增加数据集. 例如,Wang等[63]利用自然语言处理流程对高温合金的化学成分、属性数据进行了自动提取. Ren等[64] 针对高通量实验技术进行腐蚀研究进行了综述,提到将高通量腐蚀实验与电化学高通量表征相结合,可以进一步提高测量的效率.
(2)利用条件分子生成模型构建与给定属性集相对应的缓蚀剂分子结构,可通过事先了解分子的SMIELS、片段(或官能团)和目标特性之间的关系,将采样限制在非常特定的分子上. 但这样可能会阻止模型发挥其潜力.
(3)要想获得具有高效率的新缓蚀剂分子,需要从新生成的分子中进行缓蚀效率预测,或者直接以与缓蚀效率相关的分子性质作为属性条件,建立条件生成模型,但目前常用于缓蚀效率预测的量子化学参数仍不能在本研究领域达成一致,将导致以已有的量子化学参数作为条件生成模型的条件输入,可能生成无法满足特定缓蚀效率的分子.
(4)生成的缓蚀剂分子需要经过实验验证其实际缓蚀性能,这样的实验验证步骤往往费时费力. 因此,如何高效地筛选和验证生成分子的实际缓蚀效果是一个挑战. 除了缓蚀效率,生成的分子还需要满足其他性质(如环保性、稳定性、成本等). 多目标优化在生成模型中的应用及其实现难度较大.
(5)生成的分子既要多样化又要具备创新性,避免生成与已有分子相似或重复的结构. 这需要模型在生成时能够探索化学空间的广度,同时保持一定的创新性. 另外,目前许多生成模型都属于“黑箱”模型,缺乏解释性. 理解模型是如何生成缓蚀剂分子的,以及生成的分子为何具有高效的缓蚀性能,对于提升模型和设计新分子具有重要意义.
虽然仍存在上述问题需要解决,但是分子生成模型在药物等研究领域的验证成功证明了其可行性,借助文本挖掘等技术整理特定腐蚀环境下的缓蚀剂分子数据集,可有效解决数据集缺失问题. 将分子生成模型和已有缓蚀剂分子筛选方法进行闭环结合,可极大缩小高效缓蚀剂分子研究的化合物空间. 另一方面,利用高通量实验和高通量计算技术,对筛选到的缓蚀剂分子进行实验验证,可进一步加速高效缓蚀剂研究.
5. 总结
本文首先总结了当前基于ML的缓蚀剂分子筛选方法,指出仅依赖缓蚀效率预测模型进行分子筛选的局限性. 利用分子生成模型生成具有某种分子特征的缓蚀剂分子,并结合分子筛选方法对生成的分子进行缓释效率预测,将大大缩小缓蚀剂化合物筛选的范围. 因此,本文进一步简要介绍了现有的缓蚀剂分子生成模型. 但由于缓蚀剂分子研究的特殊性,仍有许多问题亟待解决. 本文通过总结缓蚀剂分子发现研究中存在的问题,并展望未来的发展方向,以期为材料腐蚀研究人员提供参考.
-
![]()
图 1 ML助力缓蚀剂分子研究的思路
Figure 1. Machine learning ideas for assisting research on corrosion inhibitor molecules
![]()
图 2 面向IE预测的QSAR/QSPR建模流程
Figure 2. Quantitative structure activity/property relationship (QSAR/QSPR) modeling process for inhibition efficiency (IE) prediction
![]()
图 3 不同模型预测缓蚀剂在1 mol·L−1盐酸溶液中的缓蚀效率准确度对比图. (a) 随机森林模型; (b) 梯度增强模型; (c) 人工神经网络模型; (d) 多元线性回归模型
Figure 3. Prediction accuracies compared for corrosion inhibition efficiency of inhibitors in 1 mol·L−1 hydrochloric acid solution, using different models: (a) random forest; (b) gradient boosting; (c) artificial neural network; (d) multiple linear regression
-
[1] Hou B R, Li X G, Ma X M, et al. The cost of corrosion in China. NPJ Mater Degrad, 2017, 1: 4 doi: 10.1038/s41529-017-0005-2
[2] Finšgar M, Jackson J. Application of corrosion inhibitors for steels in acidic media for the oil and gas industry: A review. Corros Sci, 2014, 86: 17 doi: 10.1016/j.corsci.2014.04.044
[3] Fazal B R, Becker T, Kinsella B, et al. A review of plant extracts as green corrosion inhibitors for CO2 corrosion of carbon steel. NPJ Mater Degrad, 2022, 6: 5 doi: 10.1038/s41529-021-00201-5
[4] Elqars E, Oubella A, Hachim M E, et al. New 3-(2-methoxyphenyl)-isoxazole-carvone: Synthesis, spectroscopic characterization, and prevention of carbon steel corrosion in hydrochloric acid. J Mol Liq, 2022, 347: 118311 doi: 10.1016/j.molliq.2021.118311
[5] Zou Y, Wang J, Zheng Y Y. Electrochemical techniques for determining corrosion rate of rusted steel in seawater. Corros Sci, 2011, 53(1): 208 doi: 10.1016/j.corsci.2010.09.011
[6] Bahlakeh G, Ramezanzadeh B, Ramezanzadeh M. Cerium oxide nanoparticles influences on the binding and corrosion protection characteristics of a melamine-cured polyester resin on mild steel: An experimental, density functional theory and molecular dynamics simulation study. Corros Sci, 2017, 118: 69 doi: 10.1016/j.corsci.2017.01.021
[7] Boucherit L, Al-Noaimi M, Daoud D, et al. Synthesis, characterization and the inhibition activity of 3-(4-cyanophenylazo)-2, 4-pentanedione (L) on the corrosion of carbon steel, synergistic effect with other halide ions in 0.5 M H2SO4. J Mol Struct, 2019, 1177: 371 doi: 10.1016/j.molstruc.2018.09.079
[8] Gece G. The use of quantum chemical methods in corrosion inhibitor studies. Corros Sci, 2008, 50(11): 2981 doi: 10.1016/j.corsci.2008.08.043
[9] Verma D K, Aslam R, Aslam J, et al. Computational modeling: Theoretical predictive tools for designing of potential organic corrosion inhibitors. J Mol Struct, 2021, 1236: 130294 doi: 10.1016/j.molstruc.2021.130294
[10] Obot I B, MacDonald D D, Gasem Z M. Density functional theory (DFT) as a powerful tool for designing new organic corrosion inhibitors. Part 1: An overview. Corros Sci, 2015, 99: 1
[11] 文成, 田玉琬, 杨德越, 等. 智能阻锈剂LDH-NO2在钢筋混凝土中的控释机制及缓蚀性能. 工程科学学报, 2022, 44(8):1368 Wen C, Tian Y W, Yang D Y, et al. Controlled release mechanism and inhibition performance of smart inhibitor LDH-NO2 in the reinforced concrete structures. Chin J Eng, 2022, 44(8): 1368
[12] Obot I B, Gasem Z M. Theoretical evaluation of corrosion inhibition performance of some pyrazine derivatives. Corros Sci, 2014, 83: 359 doi: 10.1016/j.corsci.2014.03.008
[13] Tang Y M, Yang X Y, Yang W Z, et al. A preliminary investigation of corrosion inhibition of mild steel in 0.5 M H2SO4 by 2-amino-5-(n-pyridyl)-1, 3, 4-thiadiazole: Polarization, EIS and molecular dynamics simulations. Corros Sci, 2010, 52(5): 1801
[14] Haris N I N, Sobri S, Yusof Y A, et al. An overview of molecular dynamic simulation for corrosion inhibition of ferrous metals. Metals, 2021, 11(1): 46
[15] Chen X S, Chen Y, Cui J J, et al. Molecular dynamics simulation and DFT calculation of “green” scale and corrosion inhibitor. Comput Mater Sci, 2021, 188: 110229 doi: 10.1016/j.commatsci.2020.110229
[16] Verma C, Lgaz H, Verma D K, et al. Molecular dynamics and Monte Carlo simulations as powerful tools for study of interfacial adsorption behavior of corrosion inhibitors in aqueous phase: A review. J Mol Liq, 2018, 260: 99 doi: 10.1016/j.molliq.2018.03.045
[17] 张明, 付冬梅, 张达威, 等. 基于综合智能模型的碳钢大气腐蚀重要变量提取和依赖关系挖掘. 工程科学学报, 2023, 45(3):407 Zhang M, Fu D M, Zhang D W, et al. Extraction of important variables and mining of dependencies of atmospheric corrosion of carbon steel based on a comprehensive intelligent model. Chin J Eng, 2023, 45(3): 407
[18] 尹志彪, 王莎莎, 祝振洪, 等. 北京地区土壤腐蚀性关键参量与Q235钢腐蚀速率预测模型研究. 工程科学学报, 2023, 45(11):1939 Yin Z B, Wang S S, Zhu Z H, et al. Key parameters of soil corrosivity and a model for predicting the corrosion rate of Q235steel in Beijing. Chin J Eng, 2023, 45(11): 1939
[19] Fernandez M, Breedon M, Cole I S, et al. Modeling corrosion inhibition efficacy of small organic molecules as non-toxic chromate alternatives using comparative molecular surface analysis (CoMSA). Chemosphere, 2016, 160: 80 doi: 10.1016/j.chemosphere.2016.06.044
[20] Gutiérrez E, Rodríguez J A, Cruz-Borbolla J, et al. Development of a predictive model for corrosion inhibition of carbon steel by imidazole and benzimidazole derivatives. Corros Sci, 2016, 108: 23 doi: 10.1016/j.corsci.2016.02.036
[21] Camacho-Mendoza R L, Feria L, Zárate-Hernández L Á, et al. New QSPR model for prediction of corrosion inhibition using conceptual density functional theory. J Mol Model, 2022, 28(8): 238 doi: 10.1007/s00894-022-05240-6
[22] Winkler D. Predicting the performance of organic corrosion inhibitors. Metals, 2017, 7(12): 553 doi: 10.3390/met7120553
[23] Ser C T, Žuvela P, Wong M W. Prediction of corrosion inhibition efficiency of pyridines and quinolines on an iron surface using machine learning-powered quantitative structure-property relationships. Appl Surf Sci, 2020, 512: 145612 doi: 10.1016/j.apsusc.2020.145612
[24] Quadri T W, Olasunkanmi L O, Akpan E D, et al. Development of QSAR-based (MLR/ANN) predictive models for effective design of pyridazine corrosion inhibitors. Mater Today Commun, 2022, 30: 103163 doi: 10.1016/j.mtcomm.2022.103163
[25] Quadri T W, Olasunkanmi L O, Fayemi O E, et al. Multilayer perceptron neural network-based QSAR models for the assessment and prediction of corrosion inhibition performances of ionic liquids. Comput Mater Sci, 2022, 214: 111753 doi: 10.1016/j.commatsci.2022.111753
[26] Du L, Zhao H X, Hu H X, et al. Quantum chemical and molecular dynamics studies of imidazoline derivatives as corrosion inhibitor and quantitative structure–activity relationship (QSAR) analysis using the support vector machine (SVM) method. J Theor Comput Chem, 2014, 13(2): 1450012 doi: 10.1142/S0219633614500126
[27] Zhao H X, Zhang X H, Ji L, et al. Quantitative structure–activity relationship model for amino acids as corrosion inhibitors based on the support vector machine and molecular design. Corros Sci, 2014, 83: 261 doi: 10.1016/j.corsci.2014.02.023
[28] Alamri A H, Alhazmi N. Development of data driven machine learning models for the prediction and design of pyrimidine corrosion inhibitors. J Saudi Chem Soc, 2022, 26(6): 101536 doi: 10.1016/j.jscs.2022.101536
[29] Sutojo T, Rustad S, Akrom M, et al. A machine learning approach for corrosion small datasets. NPJ Mater Degrad, 2023, 7: 18 doi: 10.1038/s41529-023-00336-7
[30] Galvão T L P, Novell-Leruth G, Kuznetsova A, et al. Elucidating structure–property relationships in aluminum alloy corrosion inhibitors by machine learning. J Phys Chem C, 2020, 124(10): 5624 doi: 10.1021/acs.jpcc.9b09538
[31] Akrom M, Rustad S, Dipojono H K. A machine learning approach to predict the efficiency of corrosion inhibition by natural product-based organic inhibitors. Phys Scr, 2024, 99(3): 036006 doi: 10.1088/1402-4896/ad28a9
[32] Pham T H, Le P K, Son D N. A data-driven QSPR model for screening organic corrosion inhibitors for carbon steel using machine learning techniques. RSC Adv, 2024, 14(16): 11157 doi: 10.1039/D4RA02159B
[33] Kokalj A, Lozinšek M, Kapun B, et al. Simplistic correlations between molecular electronic properties and inhibition efficiencies: Do they really exist? Corros Sci, 2021, 179: 108856
[34] Kokalj A. Molecular modeling of organic corrosion inhibitors: Calculations, pitfalls, and conceptualization of molecule–surface bonding. Corros Sci, 2021, 193: 109650 doi: 10.1016/j.corsci.2021.109650
[35] Dai J X, Fu D M, Song G X, et al. Cross-category prediction of corrosion inhibitor performance based on molecular graph structures via a three-level message passing neural network model. Corros Sci, 2022, 209: 110780 doi: 10.1016/j.corsci.2022.110780
[36] Ma J B, Dai J X, Guo X, et al. Data-driven corrosion inhibition efficiency prediction model incorporating 2D–3D molecular graphs and inhibitor concentration. Corros Sci, 2023, 222: 111420 doi: 10.1016/j.corsci.2023.111420
[37] Landrum G. Rdkit documentation. Release, 2013, 1: 4
[38] Mauri A, Consonni V, Pavan M, et al. Dragon software: An easy approach to molecular descriptor calculations. Match, 2006, 56(2): 237
[39] Yap C W. PaDEL-descriptor: An open source software to calculate molecular descriptors and fingerprints. J Comput Chem, 2011, 32(7): 1466 doi: 10.1002/jcc.21707
[40] Katritzky A R, Perumal S, Petrukhin R, et al. Codessa-based theoretical QSPR model for hydantoin HPLC-RT lipophilicities. J Chem Inf Comput Sci, 2001, 41(3): 569 doi: 10.1021/ci000099t
[41] Hadisaputra S, Irham A D, Purwoko A A, et al. Development of QSPR models for furan derivatives as corrosion inhibitors for mild steel. Int J Electrochem Sci, 2023, 18(8): 100207 doi: 10.1016/j.ijoes.2023.100207
[42] Rajan K. Informatics for Materials Science and Engineering: Data-Driven Discovery for Accelerated Experimentation and Application. Oxford: Butterworth-Heinemann, 2013.
[43] Cheng Z Y, Zou C M, Dong J W, et al. Outlier detection using isolation forest and local outlier factor // Proceedings of the Conference on Research in Adaptive and Convergent Systems. Chongqing, 2019: 161
[44] Khaled K F. Modeling corrosion inhibition of iron in acid medium by genetic function approximation method: A QSAR model. Corros Sci, 2011, 53(11): 3457 doi: 10.1016/j.corsci.2011.01.035
[45] Chafai N, Salhi H, Hadjira A, et al. Development of new models to predict the corrosion inhibition efficiency as functions of some molecular descriptors using statistical analysis. J Indian Chem Soc, 2023, 100(9): 101073 doi: 10.1016/j.jics.2023.101073
[46] Quadri T W, Olasunkanmi L O, Fayemi O E, et al. Computational insights into quinoxaline-based corrosion inhibitors of steel in HCl: Quantum chemical analysis and QSPR-ANN studies. Arab J Chem, 2022, 15(7): 103870 doi: 10.1016/j.arabjc.2022.103870
[47] Quadri T W, Olasunkanmi L O, Fayemi O E, et al. Predicting protection capacities of pyrimidine-based corrosion inhibitors for mild steel/HCl interface using linear and nonlinear QSPR models. J Mol Model, 2022, 28(9): 254 doi: 10.1007/s00894-022-05245-1
[48] Iyer R S, Iyer N S, Rugmini Ammal P, et al. Harnessing machine learning and virtual sample generation for corrosion studies of 2-alkyl benzimidazole scaffold small dataset with an experimental validation. J Mol Struct, 2024, 1306: 137767 doi: 10.1016/j.molstruc.2024.137767
[49] Akrom M, Rustad S, Saputro A G, et al. Data-driven investigation to model the corrosion inhibition efficiency of Pyrimidine-Pyrazole hybrid corrosion inhibitors. Comput Theor Chem, 2023, 1229: 114307 doi: 10.1016/j.comptc.2023.114307
[50] Akrom M, Rustad S, Saputro A G, et al. A combination of machine learning model and density functional theory method to predict corrosion inhibition performance of new diazine derivative compounds. Mater Today Commun, 2023, 35: 106402 doi: 10.1016/j.mtcomm.2023.106402
[51] Gong H Y, Fu Z H, Ma L W, et al. Inhibitor_Mol_VAE: A variational autoencoder approach for generating corrosion inhibitor molecules. NPJ Mater Degrad, 2024, 8: 102 doi: 10.1038/s41529-024-00518-x
[52] Weininger D. SMILES, a chemical language and information system. 1. introduction to methodology and encoding rules. J Chem Inf Comput Sci, 1988, 28(1): 31
[53] Hao Z K, Lu C Q, Huang Z Y, et al. ASGN: An active semi-supervised graph neural network for molecular property prediction // Proceedings of the 26th ACM SIGKDD International Conference on Knowledge Discovery & Data Mining. Virtual Event, 2020: 731
[54] Asif N A, Sarker Y, Chakrabortty R K, et al. Graph neural network: A comprehensive review on non-euclidean space. IEEE Access, 2021, 9: 60588 doi: 10.1109/ACCESS.2021.3071274
[55] Wu Z Q, Ramsundar B, Feinberg E N, et al. MoleculeNet: A benchmark for molecular machine learning. Chem Sci, 2017, 9(2): 513
[56] Yang K, Swanson K, Jin W G, et al. Analyzing learned molecular representations for property prediction. J Chem Inf Model, 2019, 59(8): 3370 doi: 10.1021/acs.jcim.9b00237
[57] Gilmer J, Schoenholz S S, Riley P F, et al. Neural message passing for quantum chemistry // Proceedings of the 34th International Conference on Machine Learning, PMLR 70. Sydney, 2017: 1263
[58] Wang X F, Li Z, Jiang M J, et al. Molecule property prediction based on spatial graph embedding. J Chem Inf Model, 2019, 59(9): 3817 doi: 10.1021/acs.jcim.9b00410
[59] Withnall M, Lindelöf E, Engkvist O, et al. Building attention and edge message passing neural networks for bioactivity and physical–chemical property prediction. J Cheminf, 2020, 12(1): 1 doi: 10.1186/s13321-019-0407-y
[60] Winkler D A, Breedon M, Hughes A E, et al. Towards chromate-free corrosion inhibitors: Structure–property models for organic alternatives. Green Chem, 2014, 16(6): 3349 doi: 10.1039/C3GC42540A
[61] Zhavoronkov A, Ivanenkov Y A, Aliper A, et al. Deep learning enables rapid identification of potent DDR1 kinase inhibitors. Nat Biotechnol, 2019, 37(9): 1038 doi: 10.1038/s41587-019-0224-x
[62] Ren F, Ding X, Zheng M, et al. AlphaFold accelerates artificial intelligence powered drug discovery: Efficient discovery of a novel CDK20 small molecule inhibitor. Chem Sci, 2023, 14(6): 1443 doi: 10.1039/D2SC05709C
[63] Wang W R, Jiang X, Tian S H, et al. Automated pipeline for superalloy data by text mining. NPJ Comput Mater, 2022, 8: 9 doi: 10.1038/s41524-021-00687-2
[64] Ren C H, Ma L W, Zhang D W, et al. High-throughput experimental techniques for corrosion research: A review. Mater Genome Eng Adv, 2023, 1(2): e20 doi: 10.1002/mgea.20
 下载:
下载:
计量
- 文章访问数: 219
- HTML全文浏览量: 19
- PDF下载量: 46
